Ewald summation
Ewald summation, named after Paul Peter Ewald, is a method for computing the interaction energies of periodic systems (e.g. crystals), particularly electrostatic energies. Ewald summation is a special case of the Poisson summation formula, replacing the summation of interaction energies in real space with an equivalent summation in Fourier space. The advantage of this approach is the rapid convergence of the Fourier-space summation compared to its real-space equivalent when the real-space interactions are long-range. Because electrostatic energies consist of both short- and long-range interactions, it is maximally efficient to decompose the interaction potential into a short-range component summed in real space and a long-range component summed in Fourier space.
Contents |
Derivation
Ewald summation rewrites the interaction potential as the sum of two terms
where 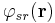 represents the short-range term that sums quickly in real space and
represents the short-range term that sums quickly in real space and 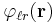 represents the long-range term that sums quickly in Fourier space. The long-ranged part should be finite for all arguments (most notably r = 0) but may have any convenient mathematical form, most typically a Gaussian distribution. The method assumes that the short-range part can be summed easily; hence, the problem becomes the summation of the long-range term. Due to the use of the Fourier sum, the method implicitly assumes that the system under study is infinitely periodic (a sensible assumption for the interiors of crystals). One repeating unit of this hypothetical periodic system is called a unit cell. One such cell is chosen as the "central cell" for reference and the remaining cells are called images.
represents the long-range term that sums quickly in Fourier space. The long-ranged part should be finite for all arguments (most notably r = 0) but may have any convenient mathematical form, most typically a Gaussian distribution. The method assumes that the short-range part can be summed easily; hence, the problem becomes the summation of the long-range term. Due to the use of the Fourier sum, the method implicitly assumes that the system under study is infinitely periodic (a sensible assumption for the interiors of crystals). One repeating unit of this hypothetical periodic system is called a unit cell. One such cell is chosen as the "central cell" for reference and the remaining cells are called images.
The long-range interaction energy is the sum of interaction energies between the charges of a central unit cell and all the charges of the lattice. Hence, it can be represented as a double integral over two charge density fields representing the fields of the unit cell and the crystal lattice
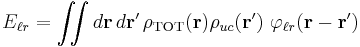
where the unit-cell charge density field 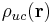 is a sum over the positions
is a sum over the positions 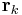 of the charges
of the charges 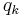 in the central unit cell
in the central unit cell
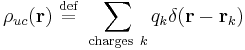
and the total charge density field 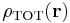 is the same sum over the unit-cell charges
is the same sum over the unit-cell charges 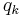 and their periodic images
and their periodic images
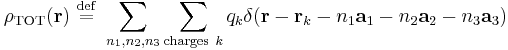
Here, 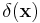 is the Dirac delta function,
is the Dirac delta function, 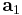 ,
, 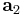 and
and 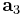 are the lattice vectors and
are the lattice vectors and 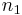 ,
, 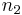 and
and 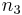 range over all integers. The total field can be represented as a convolution of with a lattice function
range over all integers. The total field can be represented as a convolution of with a lattice function 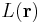
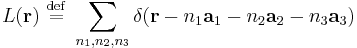
Since this is a convolution, the Fourier transformation of is a product
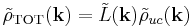
where the Fourier transform of the lattice function is another sum over delta functions
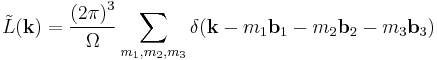
where the reciprocal space vectors are defined 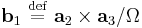 (and cyclic permutations) where
(and cyclic permutations) where 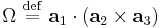 is the volume of the central unit cell (if it is geometrically a parallelepiped, which is often but not necessarily the case). Note that both and
is the volume of the central unit cell (if it is geometrically a parallelepiped, which is often but not necessarily the case). Note that both and 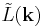 are real, even functions.
are real, even functions.
For brevity, define an effective single-particle potential
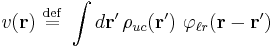
Since this is also a convolution, the Fourier transformation of the same equation is a product
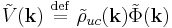
where the Fourier transform is defined
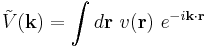
The energy can now be written as a single field integral
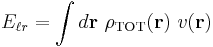
Using Parseval's theorem, the energy can also be summed in Fourier space
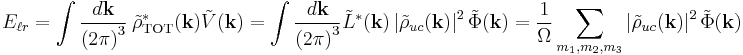
where 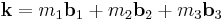 in the final summation.
in the final summation.
This is the essential result. Once 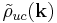 is calculated, the summation/integration over
is calculated, the summation/integration over 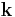 is straightforward and should converge quickly. The most common reason for lack of convergence is a poorly defined unit cell, which must be charge neutral to avoid infinite sums.
is straightforward and should converge quickly. The most common reason for lack of convergence is a poorly defined unit cell, which must be charge neutral to avoid infinite sums.
Particle mesh Ewald (PME) method
Ewald summation was developed as a method of theoretical physics, long before the advent of computers. However, the Ewald method has enjoyed widespread use since the 1970s in computer simulations of particle systems, especially those interacting via an inverse square force law such as gravity or electrostatics. Applications include simulations of plasmas, galaxies and molecules.
As in normal Ewald summation, a generic interaction potential is separated into two terms 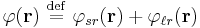 - a short-ranged part that sums quickly in real space and a long-ranged part that sums quickly in Fourier space. The basic idea of particle mesh Ewald summation is to replace the direct summation of interaction energies between point particles
- a short-ranged part that sums quickly in real space and a long-ranged part that sums quickly in Fourier space. The basic idea of particle mesh Ewald summation is to replace the direct summation of interaction energies between point particles
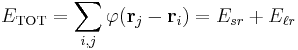
with two summations, a direct sum 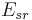 of the short-ranged potential in real space
of the short-ranged potential in real space
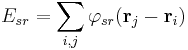
(that is the particle part of particle mesh Ewald) and a summation in Fourier space of the long-ranged part
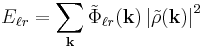
where 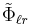 and
and 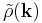 represent the Fourier transforms of the potential and the charge density (that's the Ewald part). Since both summations converge quickly in their respective spaces (real and Fourier), they may be truncated with little loss of accuracy and great improvement in required computational time. To evaluate the Fourier transform of the charge density field efficiently, one uses the Fast Fourier transform, which requires that the density field be evaluated on a discrete lattice in space (that's the mesh part).
represent the Fourier transforms of the potential and the charge density (that's the Ewald part). Since both summations converge quickly in their respective spaces (real and Fourier), they may be truncated with little loss of accuracy and great improvement in required computational time. To evaluate the Fourier transform of the charge density field efficiently, one uses the Fast Fourier transform, which requires that the density field be evaluated on a discrete lattice in space (that's the mesh part).
Due to the periodicity assumption implicit in Ewald summation, applications of the PME method to physical systems require the imposition of periodic symmetry. Thus, the method is best suited to systems that can be simulated as infinite in spatial extent. In molecular dynamics simulations this is normally accomplished by deliberately constructing a charge-neutral unit cell that can be infinitely "tiled" to form images; however, to properly account for the effects of this approximation, these images are reincorporated back into the original simulation cell. The overall effect is called a periodic boundary condition. To visualize this most clearly, think of a unit cube; the upper face is effectively in contact with the lower face, the right with the left face, and the front with the back face. As a result the unit cell size must be carefully chosen to be large enough to avoid improper motion correlations between two faces "in contact", but still small enough to be computationally feasible. The definition of the cutoff between short- and long-range interactions can also introduce artifacts.
The restriction of the density field to a mesh makes the PME method more efficient for systems with "smooth" variations in density, or continuous potential functions. Localized systems or those with large fluctuations in density may be treated more efficiently with the fast multipole method of Greengard and Rokhlin.
Dipole term
The electrostatic energy of a polar crystal (i.e., a crystal with a net dipole 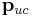 in the unit cell) is conditionally convergent, i.e., depends on the order of the summation. For example, if the dipole-dipole interactions of a central unit cell with unit cells located on an ever-increasing cube, the energy converges to a different value than if the interaction energies had been summed spherically. Roughly speaking, this conditional convergence arises because (1) the number of interacting dipoles on a shell of radius
in the unit cell) is conditionally convergent, i.e., depends on the order of the summation. For example, if the dipole-dipole interactions of a central unit cell with unit cells located on an ever-increasing cube, the energy converges to a different value than if the interaction energies had been summed spherically. Roughly speaking, this conditional convergence arises because (1) the number of interacting dipoles on a shell of radius 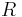 grows like
grows like 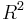 ; (2) the strength of a single dipole-dipole interaction falls like
; (2) the strength of a single dipole-dipole interaction falls like 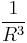 ; and (3) the mathematical summation
; and (3) the mathematical summation 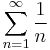 diverges.
diverges.
This somewhat surprising result can be reconciled with the finite energy of real crystals because such crystals are not infinite, i.e., have a particular boundary. More specifically, the boundary of a polar crystal has an effective surface charge density on its surface 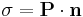 where
where 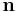 is the surface normal vector and
is the surface normal vector and 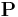 represents the net dipole moment per volume. The interaction energy
represents the net dipole moment per volume. The interaction energy 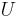 of the dipole in a central unit cell with that surface charge density can be written
of the dipole in a central unit cell with that surface charge density can be written
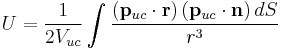
where and 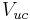 are the net dipole moment and volume of the unit cell,
are the net dipole moment and volume of the unit cell, 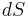 is an infinitesimal area on the crystal surface and
is an infinitesimal area on the crystal surface and 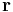 is the vector from the central unit cell to the infinitesimal area. This formula results from integrating the energy
is the vector from the central unit cell to the infinitesimal area. This formula results from integrating the energy 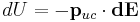 where
where 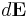 represents the infinitesimal electric field generated by an infinitesimal surface charge
represents the infinitesimal electric field generated by an infinitesimal surface charge 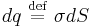 (Coulomb's law)
(Coulomb's law)
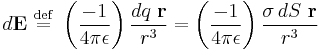
The negative sign derives from the definition of , which points towards the charge, not away from it.
History
The Ewald summation was developed by Paul Peter Ewald in 1921 (see References below) to determine the electrostatic energy (and, hence, the Madelung constant) of ionic crystals.
Scaling
Generally different Ewald summation methods give different time complexities. Direct calculation gives 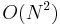 , where
, where 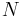 is the number of atoms in the system. The PME method gives
is the number of atoms in the system. The PME method gives 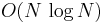 [1].
[1].
See also
References
- ^ J. Chem. Phys. 98, 10089 (1993); doi:10.1063/1.464397
- Ewald P. (1921) "Die Berechnung optischer und elektrostatischer Gitterpotentiale", Ann. Phys. 369, 253–287.
- Darden T, Perera L, Li L and Pedersen L. (1999) "New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations", Structure 7, R55–R60, doi:10.1016/S0969-2126(99)80033-1.
- Schlick T. (2002). Molecular Modeling and Simulation: An Interdisciplinary Guide Springer-Verlag Interdisciplinary Applied Mathematics, Mathematical Biology, Vol. 21. New York, NY.